Hyperkalaemia - a comprehensive(ish) guide
What does Potassium do?
Potassium is predominantly an intracellular ion, and is the main determinant of the resting membrane potential.
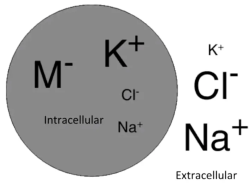
Any sort of acute change in potassium will therefore alter the resting membrane potential, and interfere with the electrical excitability of cells, thereby affecting all processes in the body that require cellular depolarisation, e.g. muscle contraction.
As with most things in physiology, acute changes outpace compensatory mechanisms, resulting in wild fluctuations, while more insidious changes are buffered and may remain clinically silent.
The normal range of serum potassium is between 3.5 to 5 mEq/L
A bit about resting membrane potential
Normal resting membrane potential is around -90mV for skeletal muscle (this may vary across cell type, e.g. -70mV for neurons).
Depolarisation = membrane potential becoming less negative (more positive)
Repolarisation = membrane potential returns to being more negative
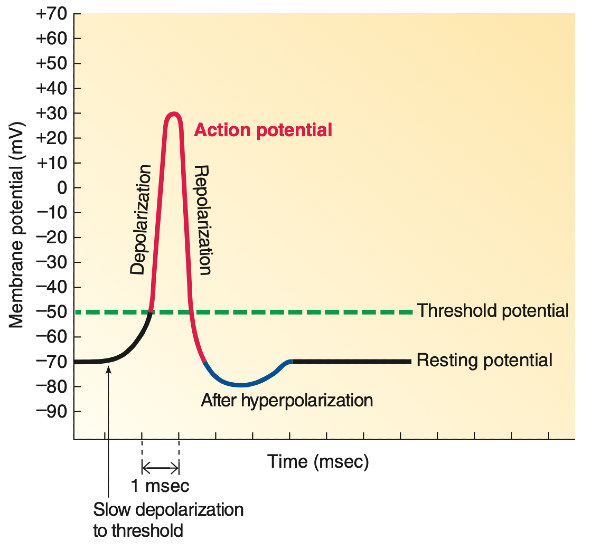
Potassium is a cation, so surely an increase in extracellular potassium would reduce the concentration gradient for K+ efflux, resulting in more positivity inside the cell, bringing the resting membrane potential closer to threshold and making the cell more excitable?
Well, yes, but not indefinitely.
Initially the reduced potassium efflux does indeed make cells more excitable as the resting potential shifts closer to threshold, so it takes less to fire an action potential.
However, eventually if extracellular potassium rises high enough, voltage gated sodium channels responsible for depolarisation enter an inactivated state. At this point, the resting membrane potential is effectively 'above' the threshold potential, meaning repolarisation to below the threshold potential can no longer occur, so future action potentials cannot be fired.
This has implications for the clinical manifestations of hyperkalaemia, discussed later.
Regulation of body potassium
To understand what can go wrong with the body's potassium balance, it would be useful to first understand the various mechanisms by which the body regulates potassium.
As a simple rule, in a single compartment system:
- Any increase in a particular thing can only be achieved by increased input or decreased output.
- Similarly any decrease in a particular thing can be achieved only by decreased input or increased output.
But when we are talking about potassium in the blood (kal-aemia), things are not so simple, because there is also the element of shift between various compartments that must be considered (we are interested mainly in shifts between the blood and the intracellular compartment).
This means we need to think about three processes, input, output, and shifts between compartments.
In terms of input of potassium, this can be through dietary intake or via other sources such as intravenous infusion. More intake increases total body potassium, while reduced intake decreases it.
Output of potassium can be via one of two options: renal or gastrointestinal.
- Renal excretion is controlled by the renin-angiotensin-aldosterone system (RAAS), which endpoint can be simplistically thought of as to increase sodium reabsorption in exchange for potassium excretion. This happens via aldosterone, the final hormone in the RAAS, which stimulates principal cells in the nephrons to absorb sodium (via ENaCs) and secrete potassium into the lumen (via ROMK). (Obviously functional kidneys are required for this to work. In a desiccated single kidney with 2 nephrons clinging on for dear life, regardless of how much aldosterone is present potassium will not be excreted as there is effectively no filtration occurring.)
- GI losses - not an innate regulatory mechanism of potassium levels, but potassium can exit the body via diarrhoea or vomiting.
Two key transporters regulate inter-compartment shifts of potassium:
- The infamous Na+/K+/ATPase, also known as the sodium potassium pump (2K+ in, 3Na+ out).
- The Na+/H+ antiporter, which indirectly affects potassium by working in conjunction with the sodium potassium pump, with Na+ as the connecting cog. Some sources simplify this and call this the H+/K+ antiporter, as the net effect is exchange of H+ for K+).
Since potassium is mostly intracellular, cell breakdown can also release potassium into the extracellular space (strictly speaking, this isn't really an inter-compartmental shift, rather loss of membrane integrity so the intracellular compartment effectively merges with the extracellular compartment).
Expanding on these three basic principles (input, output, inter-compartmental shift), the main causes of deranged body potassium levels can be derived and categorised in the next section.
Causes of Hyperkalaemia
- Increased input
- Dietary intake, e.g. potassium supplementation, i.e. KCl tablets, or a Capuchin monkey committed to a banana-only diet? (Alone unlikely to cause significant derangement due to aforementioned renal and trans-cellular regulatory mechanisms)
- Intravenous potassium infusion - typically in the setting of treatment for hypokalaemia, but an overzealous infusion may overshoot, leading to hyperkalaemia.
- Decreased output
- Chronic Kidney Disease - in fact the most common and well known cause of hyperkalaemia, as the kidneys are the main organ responsible for potassium excretion.
- Acute Kidney Injury, most commonly pre-renal where fluid volume depletion results in reduced potassium excretion secondary to reduced distal tubular water and sodium delivery.
- Gordon Syndrome: Hyperkalaemia here results from mutations in WNK1/WNK4 genes, which increase activity of the thiazide-sensitive sodium-chloride cotransporter (NCC) and decrease function of the ROMK channels in the distal nephron, leading to excessive reabsorption of sodium and chloride, while hindering potassium excretion. Effectively a type of aldosterone resistance and may be classed as a form of Type 4 RTA.
- Any form of RAAS blockade/loss of function, as the end-point of the RAAS is potassium excretion.
- Drug-induced RAAS blockade (drugs blocking RAAS system at any point)
- Potassium sparing diuretics
- ACE inhibitors
- ARBs
- NSAIDs: reduce renin secretion (via inhibition of macula densa prostaglandin production) and impair angiotensin II induced aldosterone release. Also reduces GFR via afferent arteriole vasoconstriction -> global reduction of filtration including potassium.
- Trimethoprim-sulfamethoxazole - not technically RAAS blockade, but a sort of functional aldosterone resistance. Blocks the epithelial sodium channel (ENaC) reducing sodium reabsorption, which decreases the electrochemical gradient driving potassium excretion. Interestingly increases serum creatinine without reducing GFR via blocking tubular secretion of creatinine, although this is harmless (save for causing brief clinician panic).
- Calcineurin inhibitors such as cyclosporin and tacrolimus - again not RAAS blockade, but can be again thought of as functional aldosterone resistance, in the sense that they inhibit potassium secretion via ROMK and distal sodium channel inhibition.
- Pentamidine - Similar mechanism to trimethoprim.
- Drug-induced RAAS blockade (drugs blocking RAAS system at any point)
- Endocrine disturbance mediated RAAS impairment
- Type 4 RTA (hypoaldosteronism or aldosterone resistance)
- Hyporeninaemic hypoaldosteronism (e.g. diabetes, interstitial kidney disease)
- Primary adrenal insufficiency/Addison's disease: insufficient aldosterone production
- Heparin: reduces aldosterone synthesis
- HIV: can impair adrenal function
- Aldosterone resistance (pseudohypoaldosteronism): distal nephron does not respond properly to aldosterone. Can be drug induced as mentioned previously, genetic, or structural.
- Inter-compartmental shift
- with cells still intact:
- Beta blockers - inhibit beta 2 receptors which stimulate the sodium potassium pump. Pump inhibition -> K+ stays in the blood rather than shifting into the cell.
- Digoxin -> direct inhibition of the sodium potassium pump.
- Suxamethonium induced hyperkalaemia: activation and depolarisation of nicotinic acetylcholine receptors (nAChRs) at the NMJ, leading to efflux of potassium from muscle cells. In disease states e.g. severe trauma, burns, there is increased expression of these receptors, resulting in massive dumping of potassium into the blood from myocytes.
- Insulin deficiency - insulin also stimulates the sodium potassium pump driving potassium into cells. Thus low insulin -> high serum potassium, which the kidneys then detect and excrete. So total body potassium levels may be low despite serum potassium levels being still normal or elevated.
- Acidosis - if there is excess H+ in the serum, this inhibits the Na+/H+ antiporter (usually the antiporter moves H+ out of the cell, and Na+ into the cell). The reduced intracellular Na+ reduces the sodium gradient and inhibits the Na+/K+/ATPase pump, leading to increased extracellular K+.
- Interestingly the opposite occurs where hyperkalaemia stimulates the Na/K/ATPase resulting in increased extracellular Na -> the increased sodium gradient stimulates the Na/H antiporter -> increased extracellular H+ -> acidosis
- In hyperosmolar states such as hyperglycaemic hyperosmolar syndrome or severe dehydration, water is pulled out of cells via osmosis, so the concentration of potassium in cells becomes higher. Potassium then moves down the concentration gradient into the serum.
- Hyperkalaemic periodic paralysis: The SCN4A protein controls sodium influx into skeletal muscle during contraction. Mutations cause the SCN4A channels to stay open for too long, or inactivate improperly, allowing extra sodium to be admitted into cells. This sustained depolarisation causes potassium to shift out of the skeletal muscle (via NaKATPase), resulting in hyperkalaemia. The persistent depolarisation also prevents further action potentials, leading to muscle weakness/paralysis.
- with cells not intact, i.e. cells lyse releasing contents into blood:
- Tumour lysis syndrome -> intracellular contents: uric acid, potassium, phosphate are released from cells, but phosphate binds with calcium in serum, decreasing calcium levels. So there is a constellation of hyperuricaemia, hyperkalaemia, hyperphosphataemia and HYPOcalcaemia.
- Rhabdomyolysis - breakdown of muscle cells, again release of intracellular potassium into serum
- Status epilepticus: muscle injury, similar mechanism to rhabdomyolysis.
- Haemolysis
- But this is a tricky one, because it depends on where the RBCs are destroyed.
- Only intravascular haemolysis leads to the release of potassium from RBCs directly into the serum. Examples:
- Disseminated Intravascular Coagulation - widespread clots create turbulent blood flow through vessels, increasing haemolysis.
- Thrombotic microangiopathy: extensive thromboses form in small vessels which shear RBCs as they pass through, leading to what is known as microangiopathic haemolytic anaemia (MAHA).
- Mechanical valve replacements - ongoing haemolysis due to turbulence of flow.
- In extravascular haemolysis, RBCs are destroyed in the spleen or liver, and the potassium is recycled locally so serum potassium does not rise. This explains why in sickle cell disease and thalassaemia, hyperkalaemia is unlikely.
- Artifact/pseudohyperkalaemia. This is to do with sampling error resulting from iatrogenic red cell lysis, or localised potassium shifts.
- Red cell lysis during blood collection
- Blood drawn from side of IV infusion or central line without previous flushing
- Prolonged tourniquet use
- Fist clenching during blood withdrawal
- Delayed sample analysis
- Contamination or lab error
- High cell counts - thrombocytosis (platelets have granules containing potassium), erythrocytosis, extreme leukocytosis.
Effects of Hyperkalaemia
The clinical features are non-specific, and patients are often asymptomatic. Symptoms usually appear when serum K+ reaches concentrations of >7.0mEq/L, or if there is a rapid rise in potassium levels.
As discussed before, initially cells can be hyperexcitable and patients may experience fasciculations/tingling, before the following features of decreased excitability may be seen:
- generalised muscle weakness, paralysis, paraesthesias
- respiratory distress - respiratory muscle weakness
- reduced deep tendon reflexes
- nausea, vomiting, constipation, ileus.
ECG Changes are the feature that people get the most excited about, because they are most likely to be life threatening. These include:
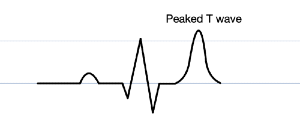
Peaked or 'tented' T waves (not the same as hyperacute T waves seen in early STEMI)
P wave flattening
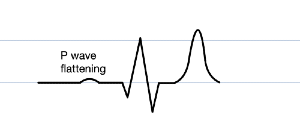
PR prolongation
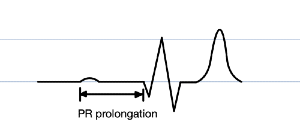
QRS widening
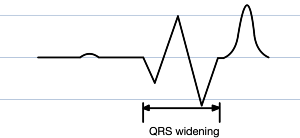
Eventually the QRS widens so much it begins to merge with the T wave, and the P wave vanishes entirely. This bizarre fusion forms what is known as a sine wave (vaguely resembling the oscillatory waveform of its mathematical namesake; why not a cosine wave simply depends on horizontal translation), which is a pre-terminal rhythm, a sign of impending doom (most commonly ventricular fibrillation or asystole)
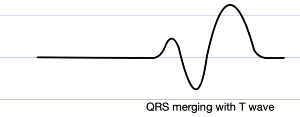
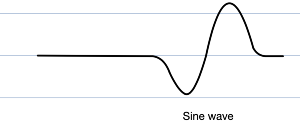
Beyond this dramatic ECG landmark, all sorts of arrhythmias, including broad complex tachycardias, AV block, and decreased cardiac contractility may ensue.
Usually these changes occur sequentially as the degree of hyperkalaemia worsens.
Workup for Hyperkalaemia
The workup for hyperkalaemia is relatively straightforward, and initially involves confirming that hyperkalaemia is actually present and not an artifact. This may be done via a repeat blood draw (to make sure the patient's arm was not subjected to strangulation with a tourniquet for half an hour before blood was drawn the first time), and platelet counts should be checked to ensure thrombocytosis is not causing pseudohyperkalaemia.
Once hyperkalaemia is confirmed, the hunt for the underlying cause can begin. This will be guided by the clinical context. Considerations may include:
- Medications: should be reviewed for common culprits such as NSAIDs, ACE inhibitors, ARBs or potassium sparing diuretics.
- Renal function: Assess creatinine, eGFR, volume status.
- Haemolysis: if haemolysis is suspected, one may search for decreased haptoglobin, anaemia, elevated unconjugated bilirubin and LDH.
- Rhabdomyolysis: Assess creatine kinase and LDH
- Acidosis: Blood gas to evaluate acid-base status.
- Tumour lysis syndrome: look for the characteristic quartet of electrolyte changes.
- Adrenal insufficiency, can look for a low aldosterone to renin ratio.
Treatment of Hyperkalaemia
Treatment depends on the degree of hyperkalaemia.
For mild hyperkalaemia (somewhat arbitrary cutoff of K+<6.0 to 6.5mEq/L) that is asymptomatic with no ECG changes, generally the management focuses on identifying and treating the underlying cause.
For severe hyperkalaemia, i.e. a hyperkalaemic emergency, e.g. K+>6.5mEq/L, presence of ECG changes, or symptoms such as muscle weakness and paralysis, management involves a combination of short-term temporising measures to rapidly shift potassium intracellularly to correct serum levels, as well as longer-term definitive measures to remove potassium from the body.
Continuous cardiac monitoring should be started immediately.
Short term temporising measures include:
- IV calcium salts:
- These increase the threshold potential of cardiac myocytes, providing a rapid cardiac stabilising effect.
- They do not actually alter serum potassium levels.
- There are 2 main options: calcium gluconate and calcium chloride. Calcium gluconate is more commonly used as it can be given via a peripheral line. Calcium chloride is 10 times more potent than calcium gluconate, so is much more effective and has faster onset, and it is also not metabolised in the liver (unlike calcium gluconate), so may be a better option with patients with liver failure. However its potency is a double edged sword because it cannot be delivered via a peripheral line, as it can cause skin necrosis, and requires central access, which may not be readily available in an emergency.
- The effect of these however, is short lived; repeat doses may be required while measures are taken to reduce potassium levels.
- Insulin:
- Stimulates the sodium potassium pump to shift potassium intracellularly.
- As a side effect there is hypoglycaemia so glucose/dextrose needs to be co-administered to maintain normoglycaemia.
- Hypoglycaemia needs to be ruled out before giving insulin, especially in the context of adrenal insufficiency, where insulin may induce a profound, refractory hypoglycaemia. (In primary adrenal insufficiency where insulin is contraindicated, the first line treatment is IV hydrocortisone).
- Thus BGLs should be measured before starting, then every 30 minutes for 2 hours, then hourly for next 4 hours.
- Salbutamol:
- Inhaled beta 2 agonists also stimulate the sodium potassium pump, causing intracellular shift of potassium.
- Has a modest effect.
- IV bicarbonate:
- Can sometimes be used if there is acidosis (the effectiveness in hyperkalaemia without acidosis is limited).
- Decreases concentration of H+ in the extracellular fluid compartment → increases intracellular Na+ via the Na+/H+ antiporter, and facilitates K+ shift into cells via sodium potassium pump (there is a bidirectional relationship between K+ and H+ levels, elevated serum K+ results in elevated serum H+ and vice versa).
- Probably works better for NAGMA.
All of the above 3 measures that reduce serum potassium levels do not actually reduce total body potassium levels. Just like when you 'clean' your room by shoving all sorts of rubbish into cupboards, these measures simply hide the excess potassium in cells.
In fact, after around 4 hours of short term effects, there will be a phenomenon of rebound hyperkalaemia, so repeat doses may be needed while waiting for more definitive measures (described next) to work.
Ultimately, you can only cram so much potassium into the metaphorical cupboard, and at some point the trash needs to be taken out. Long term, methods are needed to actually remove potassium from the body. These typically work slowly which is why the aforementioned temporising measures are required in the first place. This can be done via:
- Diuretics:
- As long as they aren't potassium sparing. Loop diuretics are commonly used, but only if the kidneys are making urine, and if the patient is sufficiently hydrated.
- Cation exchange resins, e.g. sodium polystyrene sulfonate, "Resonium":
- Bind potassium in gut and reduce absorption of potassium from diet and GI secretions.
- Comes as a white-ish powder for oral suspension. (Someone once asked the author – quite reasonably, given its physical resemblance to certain other substances, and before knowing its mechanism of action – whether it might be effective via the intranasal route. The reader may judge the efficacy of this procedure.)
- Haemodialysis - the most effective definitive therapy.
- Treating the underlying cause, e.g. correcting any volume depletion to optimise kidney function and increase delivery to tubules, or discontinuing offending medications.
References
Sherwood L. Human Physiology: From Cells to Systems. 9th ed. Boston, MA: Cengage Learning; 2015.
DiBartola SP, Autran De Morais H. Chapter 5 - Disorders of Potassium: Hypokalemia and Hyperkalemia. In: DiBartola SP, editor. Fluid, Electrolyte, and Acid-Base Disorders in Small Animal Practice (Fourth Edition). Saint Louis: W.B. Saunders; 2012. p. 92-119.